SWI-Prolog
Report approved - preliminary version for download
Thu, 2010-06-03 22:07 | by Samuel LampaMy degree project, titled "SWI-Prolog as a Semantic Web tool for semantic querying in Bioclipse" is getting closer to finish. Now my report is approved by the Scientific Reviewer (thanks, Prof. Mats Gustafsson), so I wanted to make it available here (Download PDF). Reports on typos are welcome of course! :)
Prolog query much faster when mimicking SPARQL
Sat, 2010-05-01 21:20 | by Samuel LampaI reported earlier that Jena/SPARQL outperformed Prolog for a lookup query with some numerical value comparison. It later on turned out that the results were flawed and finally that Prolog indeed was the fastest as soon as turning to datasets with more than a few hundred peaks.
The Prolog program I was using was rather complicated with recursive operations on double lists etc. Then, some week ago, I tried, in order to highlight differences in expressivity between Prolog and SPARQL, to implement a Prolog query that mimicked the structure of the SPARQL query I used, as close as possible. Interestingly it turned out that this Prolog query can be optimized to become blazing fast by reversing the order of shift values to search for, so that the largest values are searched for first. With this optimization the query outperforms both the SPARQL and the earlier used prolog code. See figure 1 below for results (The new prolog query is named "SWI-Prolog Minimal"). It appears that the querying time does not even increase with the number of triples in the RDF store!
 Figure 1: Spectrum similarity search comparison: SWI-Prolog vs. Jena
Figure 1: Spectrum similarity search comparison: SWI-Prolog vs. Jena
The explanation seems to stem from the fact that larger NMR Shift values are in general more unique than smaller values (see histogram of shift values in the full data set in figure 2 below). Thus, by testing for the largest value first, the query will be much less prone to get stuck in false leads. (Well, looking at the histogram, it appears that one could in fact do even better sorting than just from larger to smaller, like testing for values around 100 before values around 130 etc.)
 Figure 2: Histogram of NMR Shift values in 25000 spectrum dataset
Figure 2: Histogram of NMR Shift values in 25000 spectrum dataset
3rd Project Update (Integrating SWI-Prolog for Semantic Reasoning in Bioclipse)
Thu, 2010-04-08 14:17 | by Samuel LampaI just had my 3rd, and last project update presentation (before the final presenation on April 28th), presenting results from comparing the performance of the integrated SWI-Prolog against Jena and Pellet, for a spectrum similarity search query. Find the sldes below.
On larger datasets and more peaks in the query, Prolog is the fastest
Mon, 2010-03-29 16:21 | by Samuel LampaIn a previous performance test I compared a NMR Spectrum similarity search programmed in Prolog and run in SWI-Prolog integrated in Bioclipse, against a SPARQL Query doing the same task, run in Jena, and Pellet, also integrated in Bioclipse.
In that test Jena was the fastest, outperforming Prolog. The test had flaws though, as reported here with new tests added, though these new results were still not giving a clear winner.
This changed when I turned to larger datasets, testing on the range 2000-25000 spectra (where 25000 spectra corresponds to around a million RDF triples), instead of 10-100, which is a lot closer to the size of all spectra in NMRShiftDB (36419 searchable spectra as of March 29, 2010). I also used 16 instead fo 3 peak shift values in the search query. When testing with these changes, Prolog clearly took the lead.
(Bioclipse scripts, including the SPARQL Query, and the Prolog code, attached. See below).
This time I also included tests with Jena using both the in-memory RDF Store, and the Jena TDB disk based one. Interesting is that Jena run against the TDB disk based store is about double as fast as against the in-memory store! (I had to exclude the combination Pellet/TDB store, as it didn't complete for more than an hour, after which I stopped the run.)
To be noted, in these tests I did also take more measures as to avoid memory and/or caching related bias. For example, I did not rerun tests shortly after each other in a loop, but restarted Bioclipse after each run. This kind of testing took a lot more time, and I have so far only had time to do three iterataions for each specific size of dataset. Error bars, indicating the standard deviation, are included though to give an idea of the variation among the three. Hopefully I will have time to complement with a few more runs per measure point.
See figures below for results. I've included one figure where Pellet is included, and one where it is skipped, in order to get a zoom in on the Jena/Prolog difference.
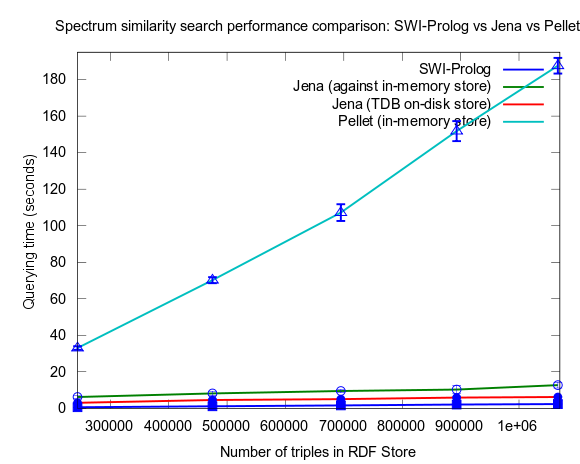
...and, Pellet excluded:
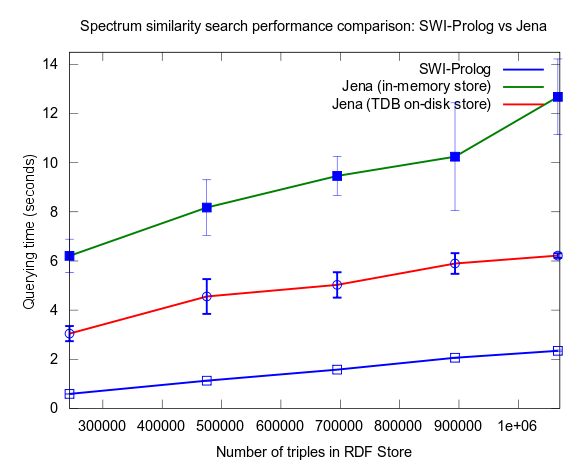
Correction of flawed results: Close competition between Jena and Prolog
Wed, 2010-03-24 04:50 | by Samuel LampaNeed to add "colon dash" before rdf_register_ns
Fri, 2010-03-12 21:17 | by Samuel LampaI could not get the rdf_db:rdf_register_ns/2 function of SWI-Prolog's semweb package to work ... getting errors like "No permission to redefine static method ..." etc.
Now I finally figured out one has to add ":-" before the line with the rdf_db:rdf_register_ns predicate, like so:
:- rdf_db:rdf_register_ns(nmr, 'http://www.nmrshiftdb.org/onto#').
or ... inside Bioclipse:
swipl.loadPrologCode(":- rdf_db:rdf_register_ns(nmr, 'http://www.nmrshiftdb.org/onto#').");Jena/SPARQL outperformed Prolog for spectrum similarity search
Thu, 2010-03-11 15:00 | by Samuel LampaUPDATE 29/3: See new results here
I was a bit worried over the performance of the RDF facilities in Bioclipse, as a SPARQL query for doing NMR Spectrum similarity search, including a numerical comparison run in Pellet (against datasets which are attached), were quite unsatisfactory, being some 2 orders of magnitude worse than some Prolog code I wrote for doing the same task (But of course, pure SPARQL with filtering is probably not what pellet is optimized for ...).
Screencast: Experimental Prolog integration in Bioclipse
Wed, 2010-03-03 20:51 | by Samuel LampaI wanted to test out some screen casting, so I chose to demo the (still experimental) SWI-Prolog integration into Bioclipse, showing how Prolog code (or a "Prolog knowledge base") can conveniently be stored inside Bioclipse's JavaScript environment (in a JS variable), loaded into the prolog engine, and then queried, all from the JS environment, and finally the results can be returned as well to the Javascript environment for further processing or output.
Note that this is still at the experimental stage, so things are a bit rough around the edges!
Performance comparison #2, Simple 13C Spectrum Similarity Search
Thu, 2009-12-03 15:53 | by Samuel LampaProlog
Bioclipse code
var start2 = new Date().getTime();
// js.say(blipkit.queryProlog( [ "findMoleculeWithPeakValuesNear", "100", "[23.3, 23.3, 23.5, 23.5, 26.1, 60.5, 90.0, 132.1, 0]", "Molecules" ] ));
js.say(blipkit.queryProlog( [ "findMoleculeWithPeakValuesNear", "100", "[12.5, 13.8, 23.8, 36.5, 44.3, 78.8, 87.3, 133.8, 0]", "Molecules" ] ));
var elapsed2 = (new Date().getTime() - start2)/1000;
js.say("Total time for finding molecule by shift values (Near-search): " + elapsed2 + " s");A usage strategy emerges
Thu, 2009-11-12 17:31 | by Samuel LampaA strategy for how to work with the Bioclipse/JPL/Prolog/Blipkit combination I'm setting up, is becoming clear.
The main idea with Bioclipse, as well as with having a prolog engine available in it, is for flexible and "interactive" knitting together of knowledge. One of the main questions regarding how to use a Bioclipse/JPL/Prolog/Blipkit combination, has been where to put the bulk of knowledge integration/reasoning code? There would in principle be three options for that:
- Bioclipse (Javascript environment)
- The Blipkit-Prolog/Bioclipse integration plugin (Java code, a.k.a. "Manager methods")
- The prolog engine (As a prolog file)
Recent blog posts
- I created a Udemy course on my favourite linux commandline productivity techniques
- My top-languages-per-use-case list
- The smallest pipeable python script
- Vote for ProcessWire to be packaged as a BitNami image
- Calling Java from Python without the JVM startup latency: NailGun and JPype
- New Google+ communities: Bioclipse, Cheminformatics, Semantic MediaWiki
- Don't use Swedish keyboard with vim/screen/bash
- Easier debugging of Go programs on the command line with CGDB
- Profiling and creating call graphs for Go programs with "go tool pprof"
- Python-like generator functions in Go