Jena/SPARQL outperformed Prolog for spectrum similarity search
UPDATE 29/3: See new results here
I was a bit worried over the performance of the RDF facilities in Bioclipse, as a SPARQL query for doing NMR Spectrum similarity search, including a numerical comparison run in Pellet (against datasets which are attached), were quite unsatisfactory, being some 2 orders of magnitude worse than some Prolog code I wrote for doing the same task (But of course, pure SPARQL with filtering is probably not what pellet is optimized for ...).
Anyway, when testing the same SPARQL query with Jena, I got some quite different results, as seen in the graph to the right; Jena in fact outperformed the prolog code! Interesting, and - I have to admit - a bit surprising ...
Test results
Pellet/SPARQL vs SWI-Prolog
So, while Prolog far outperformed Pellet for the task (at least when Pellet was to do it using SPARQL) as seen below (times in seconds) ...
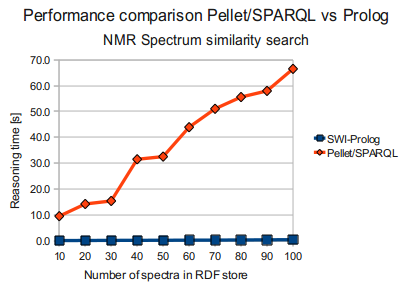
Jena/SPARQL vs SWI-Prolog
... at the same time Jena grossly outperforms Prolog, running the same SPARQL query above, as you can see below. (times in seconds)
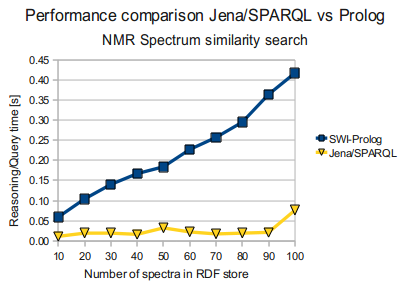
In Tabular form
| No of spectra | SWI-Prolog | Jena/SPARQL | Pellet/SPARQL |
| 10 | 0.06 | 0.01 | 9.50 |
| 20 | 0.10 | 0.02 | 14.22 |
| 30 | 0.14 | 0.02 | 15.39 |
| 40 | 0.17 | 0.02 | 31.52 |
| 50 | 0.18 | 0.03 | 32.56 |
| 60 | 0.23 | 0.02 | 43.93 |
| 70 | 0.26 | 0.02 | 51.05 |
| 80 | 0.30 | 0.02 | 55.60 |
| 90 | 0.36 | 0.02 | 57.96 |
| 100 | 0.42 | 0.08 | 66.49 |
Code
SPARQL Query
PREFIX owl: <http://www.w3.org/2002/07/owl#>
PREFIX afn: <http://jena.hpl.hp.com/ARQ/function#>
PREFIX fn: <http://www.w3.org/2005/xpath-functions#>
PREFIX nmr: <http://www.nmrshiftdb.org/onto#>
PREFIX xsd: <http://www.w3.org/2001/XMLSchema#>
PREFIX rdfs: <http://www.w3.org/2000/01/rdf-schema#>
SELECT ?s ?s1 ?s2 ?s3
WHERE {
?s nmr:hasPeak [ nmr:hasShift ?s1 ] ,
[ nmr:hasShift ?s2 ] ,
[ nmr:hasShift ?s3 ] .
FILTER ( fn:abs(?s1 - 17.6) < 0.3 &&
fn:abs(?s2 - 18.3) < 0.3 &&
fn:abs(?s3 - 22.6) < 0.3 )
} LIMIT 1"
Prolog code
% Register RDF namespaces, for use in the convenience methods at the end
:- rdf_db:rdf_register_ns(nmr, 'http://www.nmrshiftdb.org/onto#').
:- rdf_db:rdf_register_ns(xsd, 'http://www.w3.org/2001/XMLSchema#').
findMolWithPeakValsNear( SearchShiftVals, Mols ) :-
% Pick the Moleculess in 'Mol', that match the pattern:
%% listPeakShiftsOfMol( Mol, MolShiftVals ),
%% containsListElemsNear( SearchShiftVals, MolShiftVals )
% and collect them in 'Mols'.
%
% A Mol(ecule)'s shift values are collected and compared against the given
% SearchShiftVals. Then, in 'Mols', all 'Mol's, for which their shift values
% match the SearchShiftVals, are collected.
setof( Mol,
( listPeakShiftsOfMol( Mol, MolShiftVals ),
containsListElemsNear( SearchShiftVals, MolShiftVals )),
[Mols|MolTail] ).
% Given a 'Mol', give it's shiftvalues in list form, in 'ListOfPeaks'
listPeakShiftsOfMol( Mol, ListOfPeaks ) :-
hasSpectrum( Mol, Spectrum ),
findall( ShiftVal,
( hasPeak( Spectrum, Peak ),
hasShiftVal( Peak, ShiftVal ) ),
ListOfPeaks ).
% Compare two lists to see if list2 has near-matches for each value in list1
containsListElemsNear( [ElemHead|ElemTail], List ) :-
memberCloseTo( ElemHead, List ),
( containsListElemsNear( ElemTail, List );
ElemTail == [] ).
%%%%%%%%%%%%%%%%%%%%%%%%
% Recursive construct: %
%%%%%%%%%%%%%%%%%%%%%%%%
% Test first the end criterion:
memberCloseTo( X, [ Y | Tail ] ) :-
closeTo( X, Y ).
% but if the above doesn't validate, do recursively continue with the tail of List2:
memberCloseTo( X, [ Y | Tail ] ) :-
memberCloseTo( X, Tail ).
% Numerical near-match
closeTo( Val1, Val2 ) :-
abs(Val1 - Val2) =< 0.3.
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
% Convenience accessory methods %
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
hasShiftVal( Peak, ShiftVal ) :-
rdf_db:rdf( Peak, nmr:hasShift, literal(type(xsd:decimal, ShiftValLiteral))),
atom_number_create( ShiftValLiteral, ShiftVal ).
hasSpectrum( Subject, Predicate ) :-
rdf_db:rdf( Subject, nmr:hasSpectrum, Predicate).
hasPeak( Subject, Predicate ) :-
rdf_db:rdf( Subject, nmr:hasPeak, Predicate).
% Wrapper method for the atom_number/2 method which converts atoms (string
% constants) to number. The wrapper methods avoids exceptions on empty atoms,
% instead converting into a zero.
atom_number_create( Atom, Number ) :-
% IF atom is not empty
atom_length( Atom, AtomLength ), AtomLength > 0 ->
% THEN Convert the atom to a numerical value
atom_number( Atom, Number );
% ELSE Convert to a zero
atom_number( '0', Number ).
Attached files
Attached are the datasets (nmrshiftdata10...100.rdf.xml) for the different number of spectra, and the Bioclipse JavaScript files for the tests (NMR.Pellet.js, NMR.Jena.js and NMR.Swipl.js, as well as NMR.Pellet.Init, which I used for preparing the RDF stores for both Jena and Pellet).
| Attachment | Size |
|---|---|
| nmrshiftdata.10.rdf.xml | 49.83 KB |
| nmrshiftdata.20.rdf.xml | 108.37 KB |
| nmrshiftdata.30.rdf.xml | 157.07 KB |
| nmrshiftdata.40.rdf.xml | 215.49 KB |
| nmrshiftdata.50.rdf.xml | 250 KB |
| nmrshiftdata.60.rdf.xml | 308 KB |
| nmrshiftdata.70.rdf.xml | 365.74 KB |
| nmrshiftdata.80.rdf.xml | 417.52 KB |
| nmrshiftdata.90.rdf.xml | 465.25 KB |
| nmrshiftdata.100.rdf.xml | 530.94 KB |
| NMR.PelletAndJenaInit.js.txt | 5.28 KB |
| NMR.Pellet.js.txt | 2.89 KB |
| NMR.Jena_.js.txt | 3.04 KB |
| NMR.Swipl_.js.txt | 4.28 KB |
Recent blog posts
- I created a Udemy course on my favourite linux commandline productivity techniques
- My top-languages-per-use-case list
- The smallest pipeable python script
- Vote for ProcessWire to be packaged as a BitNami image
- Calling Java from Python without the JVM startup latency: NailGun and JPype
- New Google+ communities: Bioclipse, Cheminformatics, Semantic MediaWiki
- Don't use Swedish keyboard with vim/screen/bash
- Easier debugging of Go programs on the command line with CGDB
- Profiling and creating call graphs for Go programs with "go tool pprof"
- Python-like generator functions in Go